Notes on MD hands-on session of MSE6701H-CHN - Surface Energy Calculation¶
约 1227 个字 67 行代码 8 张图片 预计阅读时间 5 分钟
Content: [TOC]
0.0 Notice¶
-
请大家提前熟悉以下内容:
- 远程服务器登录(相关终端模拟器使用或
ssh命令) - 本地文件与远程服务器间的传输(相关文件传输软件如 WinSCP 或
scp/rsync命令) - Linux 基本命令
- Vim 文本编辑器
- 远程服务器登录(相关终端模拟器使用或
-
在使用课程材料前,请先阅读该目录下的
README说明文件内容来大致了解其使用说明、框架及功能,不要一开始就 “盲目” 进行操作(提交任务),以免出现不必要的错误 -
使用课程材料过程中,请熟悉课程材料的目录及文件结构!课程材料及该 Notes 均测试正常
0.1 Helpful things¶
可能有帮助的一些链接:
-
《多尺度材料模拟与计算》课程表面能计算实验材料 Gitee 仓库:MMMS-Surface-Energy-Calculation: Surface Energy Calculation Course Materials for MSE6701H Multiscale Materials Modelling and Simulation.
-
WSL 安装与使用:WSL 安装与使用 - Wiki of NES Lab
-
Linux 相关教程:LINUX-TUTORIAL
-
LAMMPS Documentation:LAMMPS Documentation
-
LAMMPS 相关教程:LAMMPS教程汇总 - lammps 加油站
-
Miniconda3 安装与使用:Miniconda3 安装与使用 - Wiki of NES Lab
-
模型构建相关开源程序
1. Preparation¶
1.1. Account lookup¶
- For the hands-on session and for your homeworks, the Siyuan cluster of HPC, SJTU will be used. You have been offered a temporary account, please click & find it here!
:::danger Note: this account is used for the course related tasks only. Any abuse will lead to suspension of your previlige to use the superclusters. :::
1.2. SJTU HPC login¶
1.2.1 Windows Setup¶
-
If your computer system is Windows, to login the Siyuan cluster, you are advised to use MobaXterm free Xserver and tabbed SSH client for Windows. The free version is good enough. Please have it installed on your computer.
-
Besides MobaXterm, you can use other terminal emulators on Windows such as Windows Terminal, Tabby, Termius and so on.
-
For your first time to run MobaXterm, please open the "Session" tab:

- which shall show you a window looks like, click on "SSH":
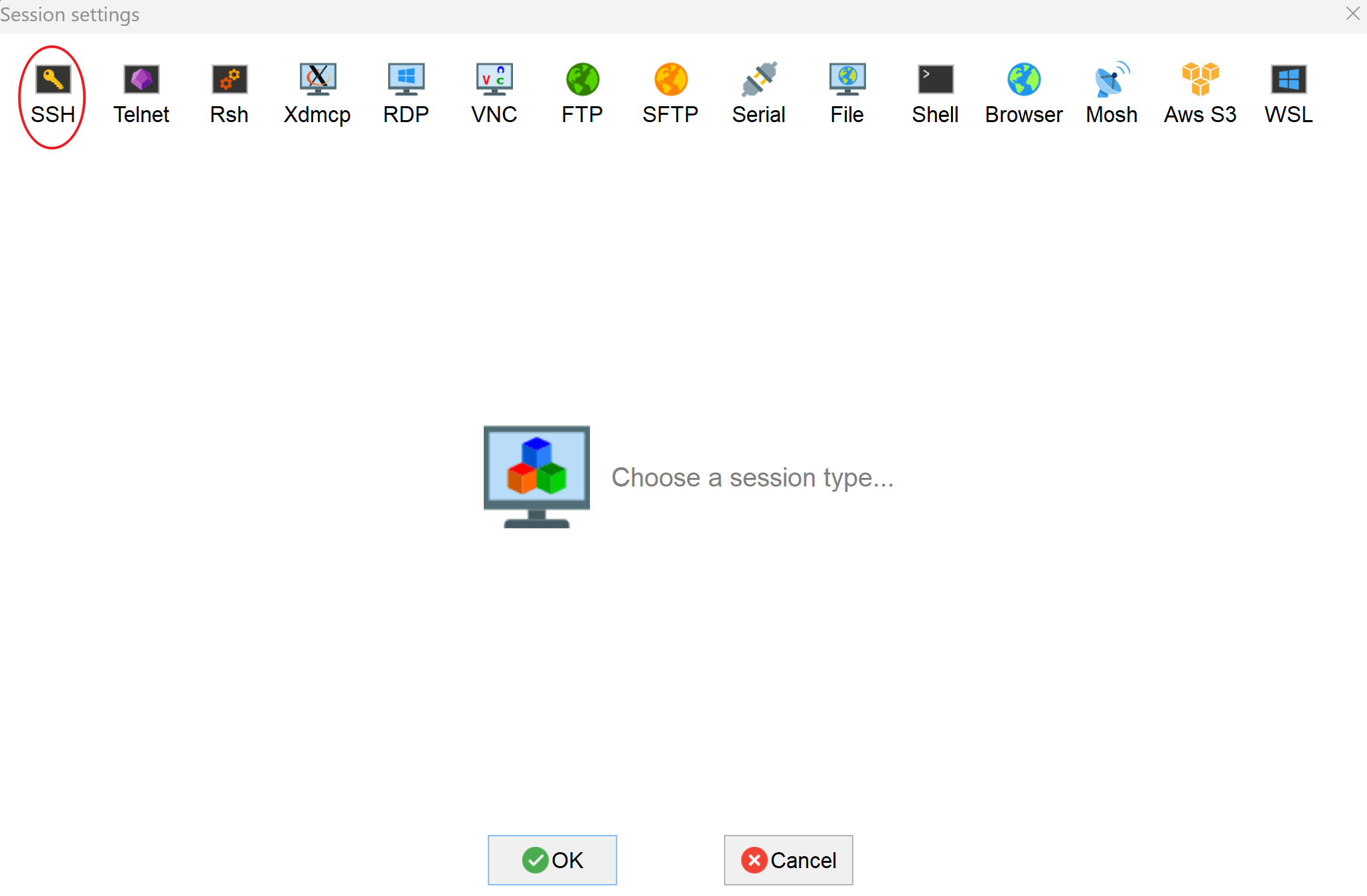
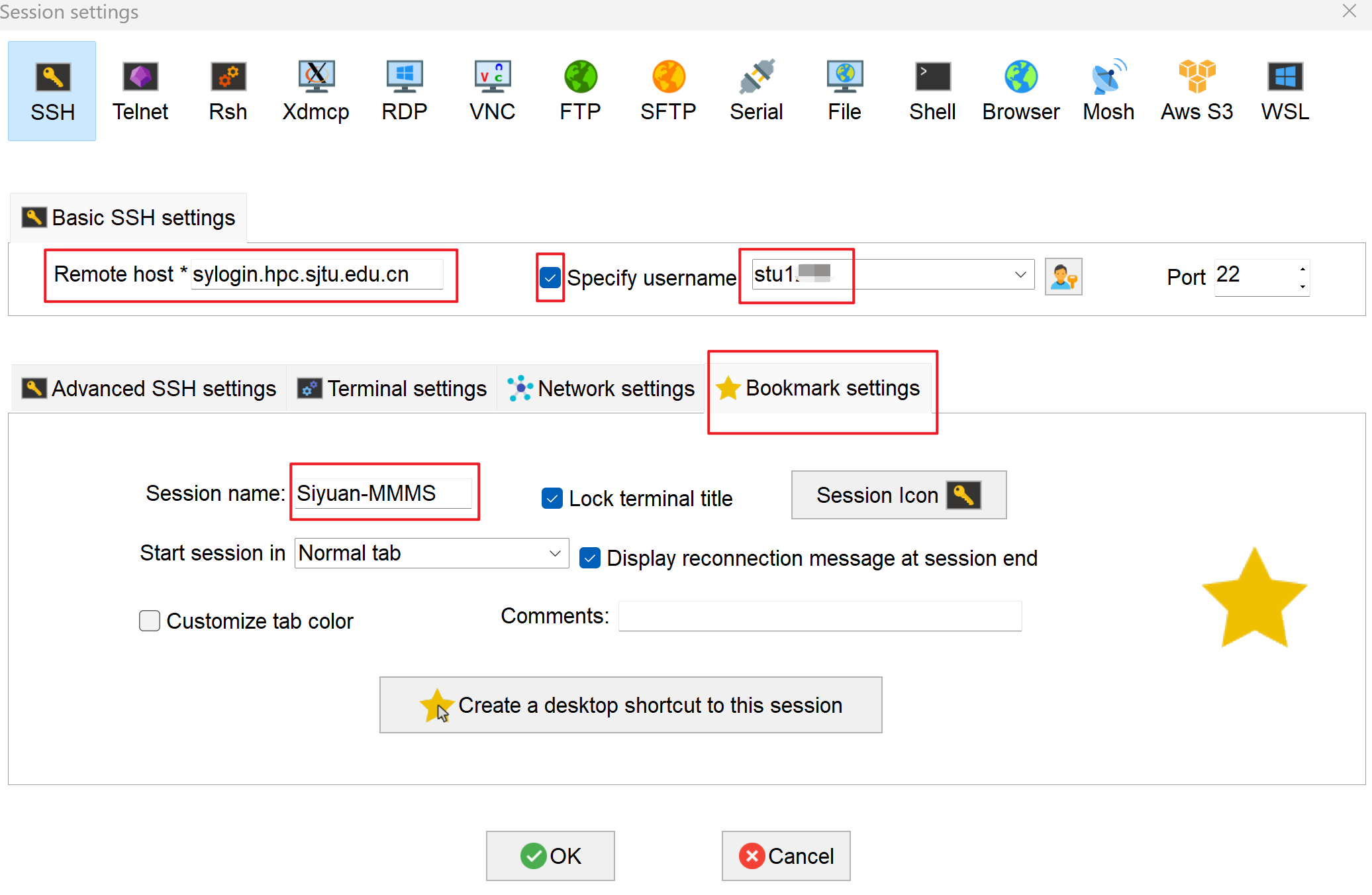
- For "Remote host", input
sylogin.hpc.sjtu.edu.cn. Check "Specify username" and input your username. Click on "Bookmark settings" and name your session as Siyuan-MMMS. Press "OK" to save:
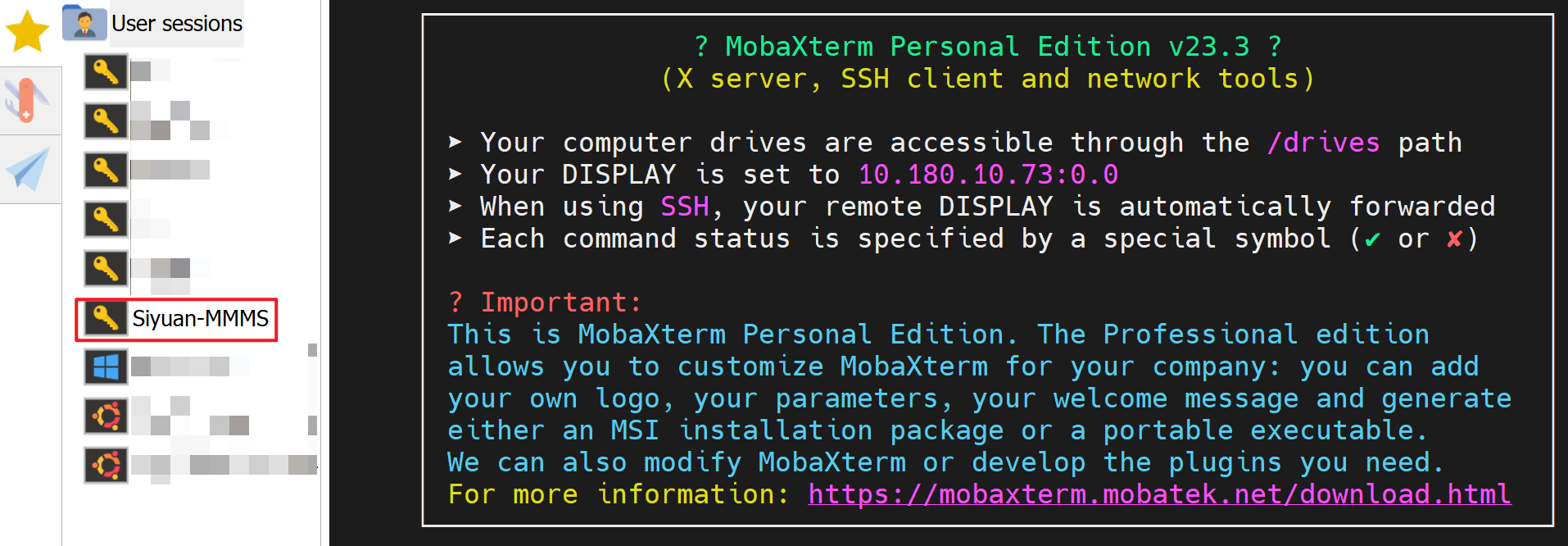
- Now you should have a bookmarked session "Siyuan-MMMS":
-
Click on "Siyuan-MMMS" to login to the remote Siyuan cluster, you will be asked for your password; and the program may ask if you would like to save your password, you can answer "yes" if you are the only person using this computer.
-
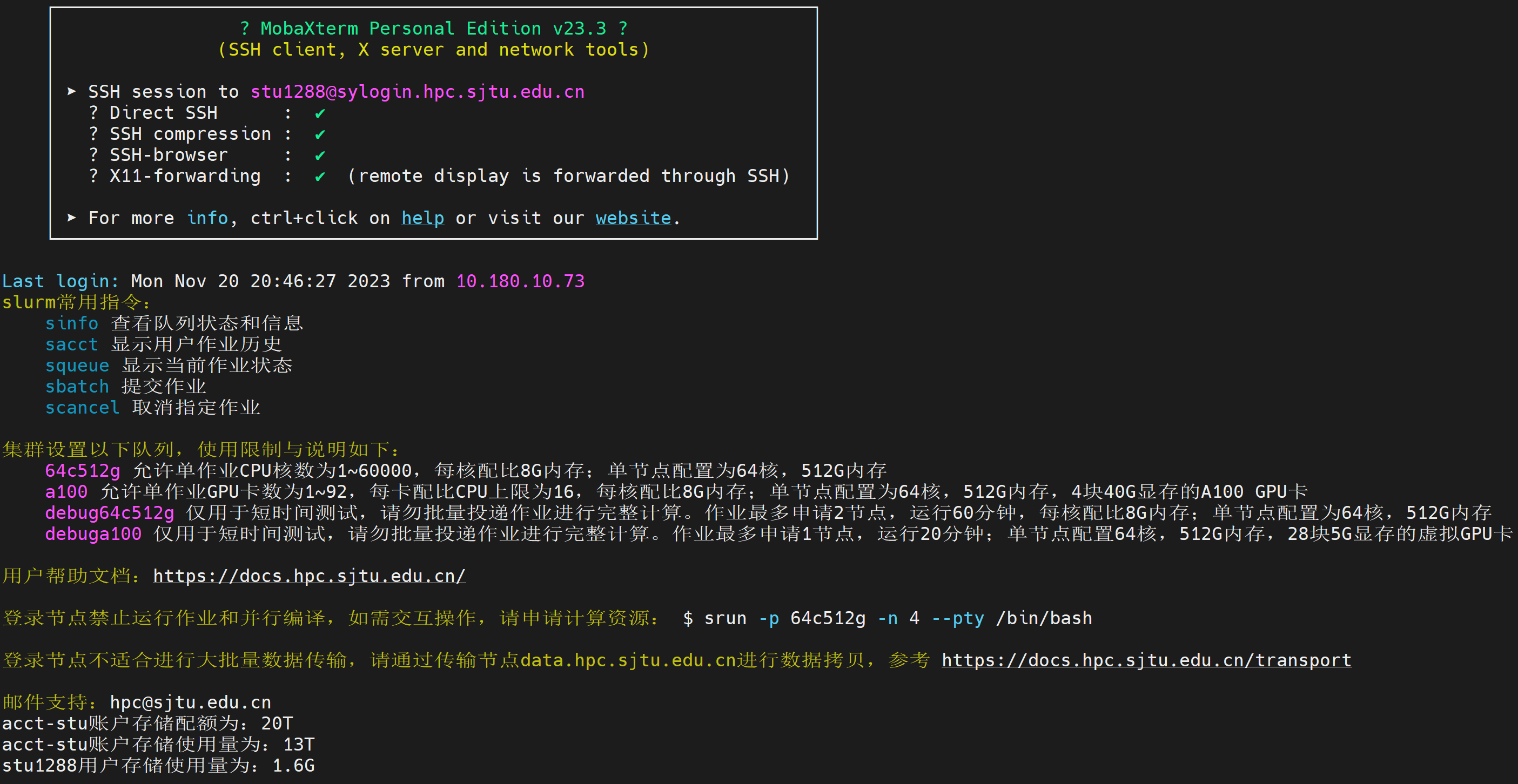
By providing the correct username and password, you should be logged into the supercluster by now:
-
To logout, you can:
- Type "exit" on the shell;
- Close MobaXterm.
-
Next time when you need to login the Siyuan cluster, simply open MobaXterm and click on "Siyuan-MMMS".
-
For method to upload/download files from the Siyuan cluster, please refer to the manual for hpc: 文件系统与数据访问 - 上海交大超算平台用户手册 Documentation
-
For submitting LAMMPS jobs on Siyuan, please refer to: LAMMPS - 上海交大超算平台用户手册 Documentation
:::danger
Note: for the jobs in this course, use at most 4 cores for your jobs. i.e., #SBATCH --ntasks-per-node=4. In most cases, 1 is good enough.
:::
1.2.2 macOS setup¶
- If your computer system is macOS, to login the Siyuan cluster, you should use SSH commands in your local terminal emulator such as:
- Terminal (macOS default App)
- iTerm2
- Termius (support macOS and Windows)
- 交我算定制版客户端Tabby - 上海交大超算平台用户手册 (support macOS and Windows)
# Note: -p 22 can be omitted
# login to Siyuan HPC
ssh -p 22 [email protected]
# Copy file from remote Siyuan to your local computer
scp -P 22 [email protected]:/dssg/home/acct-stu/stuXXX/* .
# Upload local file/folder to remote Siyuan HPC
scp -P 22 -r LocalFolderName [email protected]:/dssg/home/acct-stu/stuXXX/*
1.3. Retrieving of Course Materials¶
- Once login Siyuan, you can run the following commands on your shell, it will automatically git clone to download Course Materials
MSE6701H/MMMS-Surface-Energy-Calculationto your home directory on Siyuan.
sh -c "$(wget https://gitee.com/yangsl306/MMMS-Surface-Energy-Calculation/raw/main/mmms_sec.sh -O -)"
- If you have cloned the Course Materials, then just git pull to get latest commit.
cd ${HOME}/MSE6701H/MMMS-Surface-Energy-Calculation
current_branch=$(git branch --show-current)
git pull origin ${current_branch}
- Course Materials directory structure:
0-tools: Some tools that help to analyze the MD results.1-Bulk-Energy-Cu: Determination of equilibrium lattice constant of fcc Cu.2-Slab-Relaxation: Surface energy calculation of fcc Cu.
2. Step by step instructions on the Surface Energy Calculation¶
2.1. Determination of equilibrium lattice constant of fcc Cu¶
:::info This example illustrates the determination of the equibrium lattice constant of fcc Cu based on the adopted interatomic potential by calculating the total potential energy of the system at a series of volumes and fitting to the volume-energy data to an equation of state. :::
- Get into the directory before running the example:
- directory structure:
:::danger
There should be 5 files in this directory. One can examine the in.lmp or scan.sh to understand the method.
:::
- To run the example:
- Your job is now submitted to Siyuan for calculation. It might be waiting for the computing resource to be available. Once completed, you should find 2 more files in your directory:
- To check the content of
ev.datorlog.lammps, one can use:
- You can find the equilibrium lattice constant of fcc Cu by fitting the obtained
ev.datto Murnaghan's Equation of States.
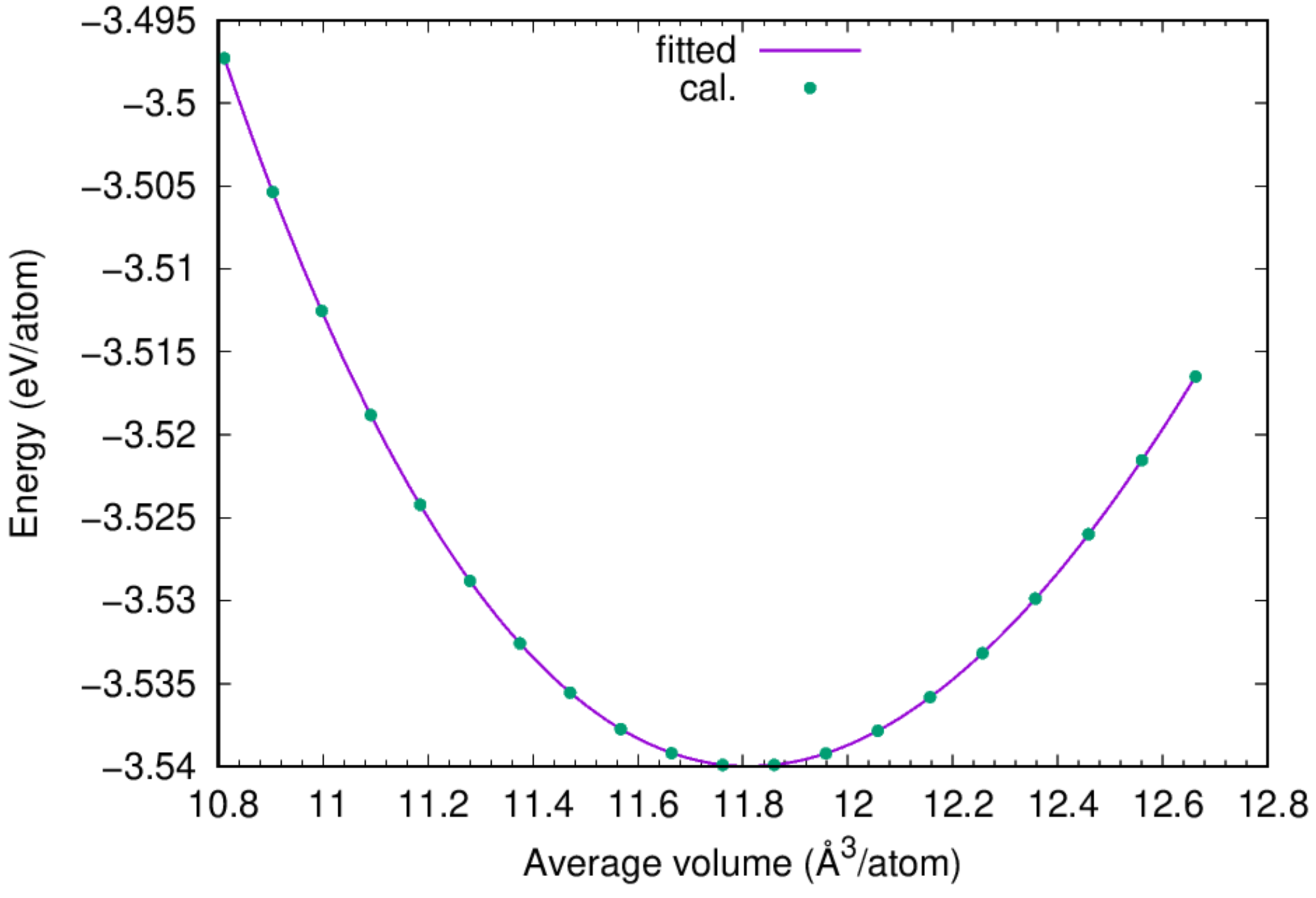
:::success According to the fitting results, the cohesive energy for fcc Cu is 3.54 eV/atom, and the lattice constant of fcc Cu based on the adopted potential is 3.615 Angstrom, agreeing perfectly with the experimental value. :::

2.3. Surface energy calculation¶
:::info The second example illustrates the calculation of the surface energy of fcc Cu(001); the equilibrium bulk energy (-3.54 eV/atom) and lattice constant 3.615 Angstrom from example (1) is used here. :::
- To run this example:
:::danger Please check the LAMMPS input files and understand the method before submitting the job: :::
- Once the job has been finished, one can find the results from the
log.lammpsfile:

:::danger
Note: OVITO can be used to visualize the atomic configuration as in dump.lammpstrj. For instruction to use ovito with Siyuan, please refer to OVITO - 上海交大超算平台用户手册 Documentation.
:::
- Use
interlayer_separations.pyPython script to measure interlayer separations of the (001) surface of fcc Cu: \(\Delta d_{12}\) =-1.05%,\(\Delta d_{23}\) =-0.33%
:::danger
Note: To use interlayer_separations.py Python script, you need to install the Python virtual environment via Miniconda3, and install the ASE, NumPy and Pandas packages. You can operate by following this blog Miniconda3 安装与使用 - Wiki of NES Lab.
:::
# Show help message
../0-tools/interlayer_separations.py -h
# Measure interlayer separations
../0-tools/interlayer_separations.py dump.lammpstrj
# Output
Inerlayer separations info:
Layer_Distance_Final Layer_Distance_Init Inerlayer_Separations Ratio(%)
16-17 1.788 1.807 -0.019 -1.05
15-16 1.802 1.808 -0.006 -0.33
14-15 1.807 1.807 0.000 0.00
13-14 1.808 1.808 0.000 0.00
12-13 1.807 1.807 0.000 0.00
11-12 1.808 1.808 0.000 0.00
10-11 1.807 1.807 0.000 0.00
9-10 1.808 1.808 0.000 0.00
8-9 1.808 1.808 0.000 0.00
7-8 1.807 1.807 0.000 0.00
6-7 1.807 1.807 0.000 0.00
5-6 1.808 1.808 0.000 0.00
4-5 1.808 1.807 0.001 0.06
3-4 1.807 1.808 -0.001 -0.06
2-3 1.802 1.807 -0.005 -0.28
1-2 1.788 1.808 -0.020 -1.11
3. Homework content & requirements¶
:::info Based on the above examples, generate Au(100), Au(110), Au(111) slab models, run minimization at 0K.
a) Calculate their surface energies at 0K;
b) Measure the interlayer separations of the surface, and compare to data in Table1 of Solid State Communications 149(37–38): 1561-1564, 2009.
c) Write a report on your experiments.
The due is: Dec 11th, 2024. :::