Notes on MD hands-on session of MSE6701H-CHN¶
约 2412 个字 129 行代码 19 张图片 预计阅读时间 10 分钟
Content: [TOC]
0. Notice¶
同学们好,今晚的课程为 MD 部分上机实操课:
- 请同学们尽量都准备好笔记本电脑并充保持充足的电量(充电器也可以带上),条件允许的同学可以把排插带上
- 本次上机实操课将涉及远程服务器登录、任务提交与文件传输,Linux 操作系统,Vim 编辑器,LAMMPS,结构建模,构型可视化及其分析等内容
- 请同学们在今晚上课前按照该链接 https://notes.sjtu.edu.cn/s/ULArhqGqt 中的操作提前安装好 Mobaxterm、Ovito、WinSCP 软件,并用自己的课程超算账号远程登录思源一号,熟悉远程登录与文件传输、Linux 基本命令、Vim 编辑器使用等基础操作
- 在使用课程材料前,请先阅读该目录下的
README文件内容来大致了解其使用说明、框架及功能,不要一开始就 “盲目” 进行操作(提交任务),以免出现不必要的错误 - 使用课程材料过程中,请熟悉课程材料的目录及文件结构!课程材料及该 Notes 均测试正常
可能有帮助的一些教程链接:
- 《多尺度材料模拟与计算》课程 MD、DFT 部分实验材料:Course Materials for MSE6701H Multiscale Materials Modelling and Simulation
- 《多尺度材料模拟与计算》课程作业相关问题:MMMS homework questions
- WSL 安装与使用:WSL 安装与使用 - Wiki of NES Lab
- Linux 相关教程:LINUX-TUTORIAL
- LAMMPS Documentation:LAMMPS documentation
- LAMMPS 相关教程:LAMMPS教程汇总 - lammps 加油站
- 模型构建相关开源程序
1. Preparation¶
1.1. Account lookup¶
- For the hands-on session and for your homeworks, the Siyuan cluster of HPC, SJTU will be used. You have been offered a temporary account, please find it here:

:::danger Note: this account is used for the course related tasks only. Any abuse will lead to suspension of your previlige to use the superclusters. :::
1.2. System login¶
-
To login the Siyuan cluster, you are advised to use MobaXterm free Xserver and tabbed SSH client for Windows. The free version is good enough. Please have it installed on your computer.
-
For your first time to run MobaXterm, please open the “Session” tab:

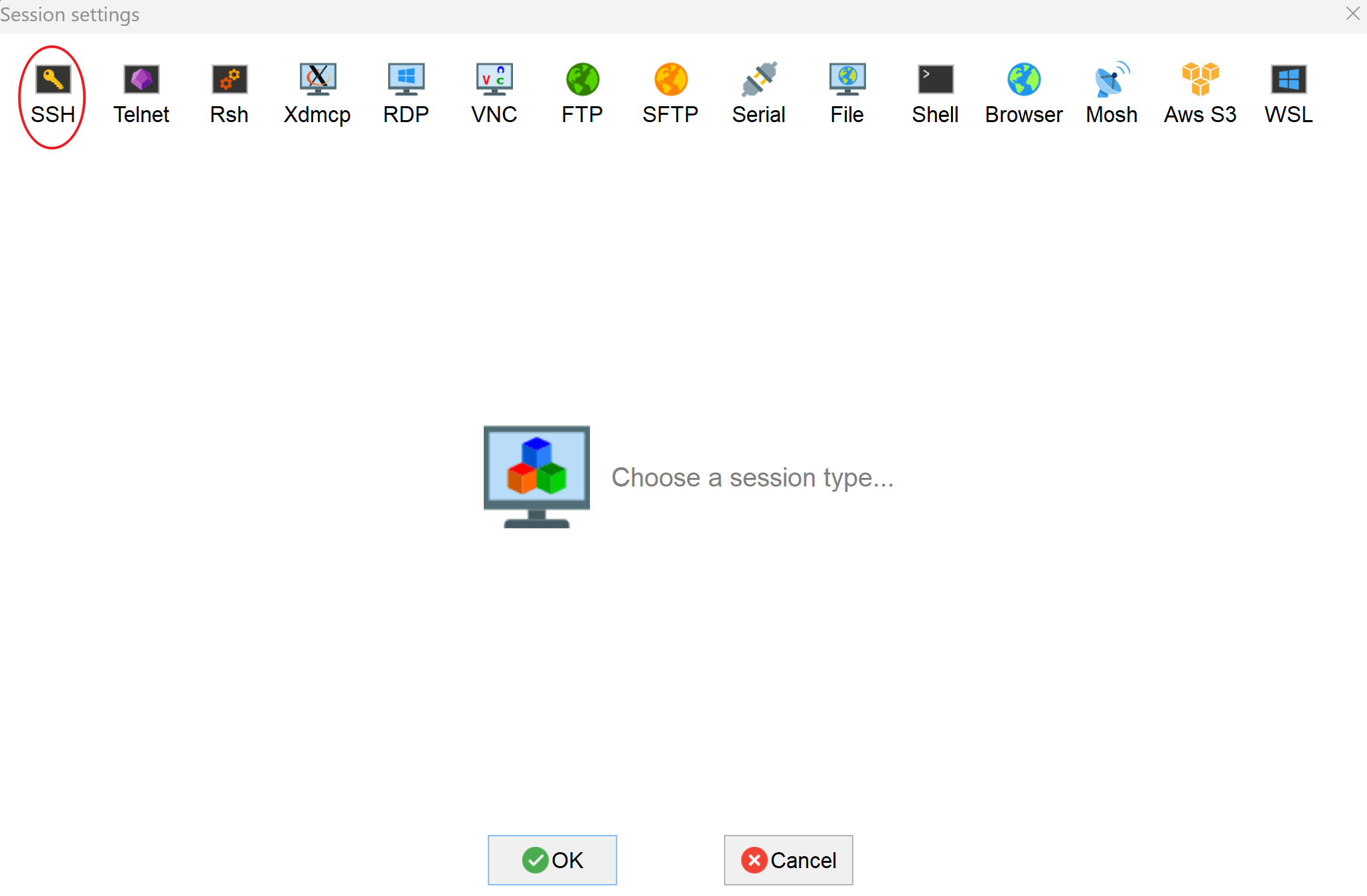
- which shall show you a window looks like, click on “SSH”:
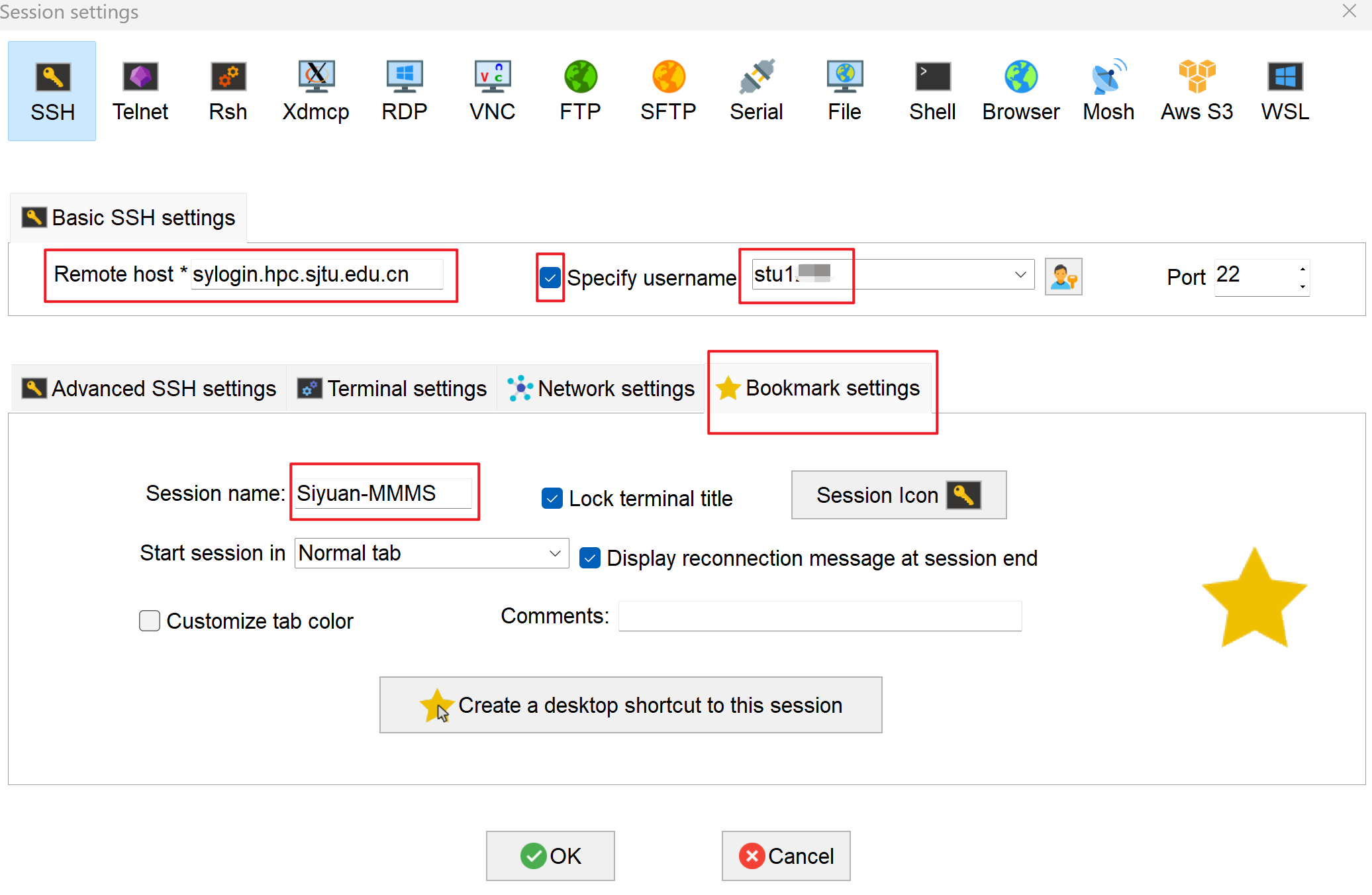
- For “Remote host”, input
sylogin.hpc.sjtu.edu.cn. Check “Specify username” and input your username. Click on “Bookmark settings” and name your session as Siyuan-MMMS. Press “OK” to save:
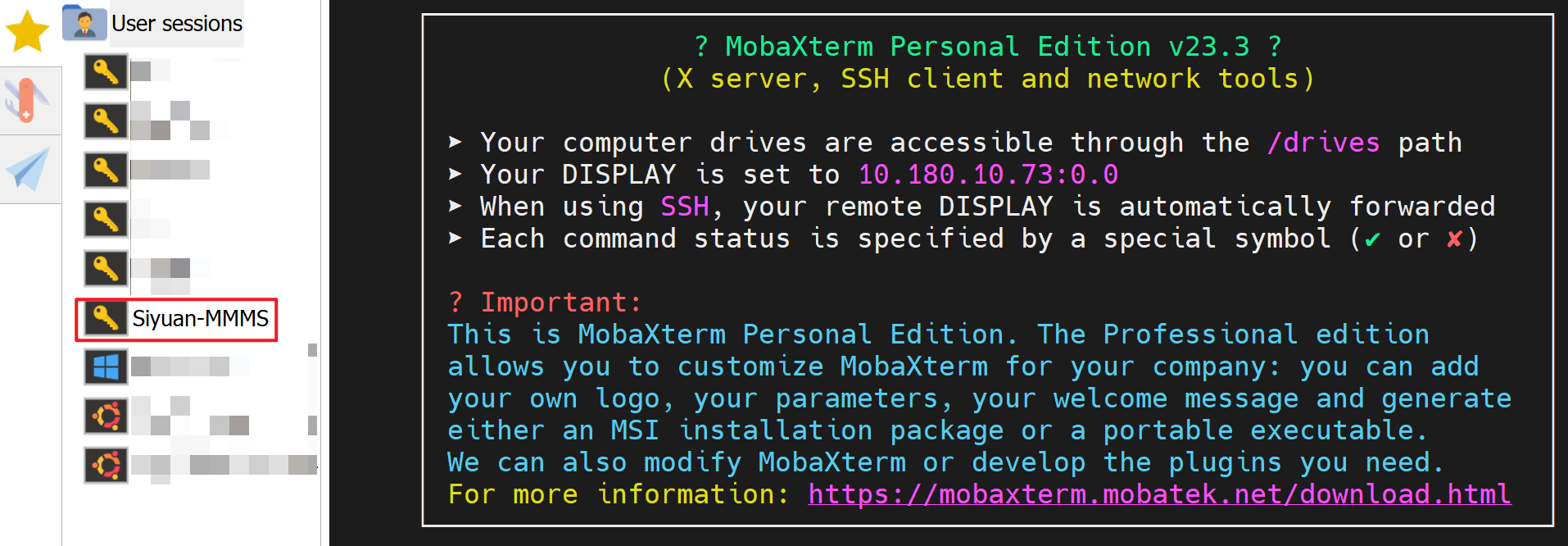
- Now you should have a bookmarked session “Siyuan-MMMS”:
-
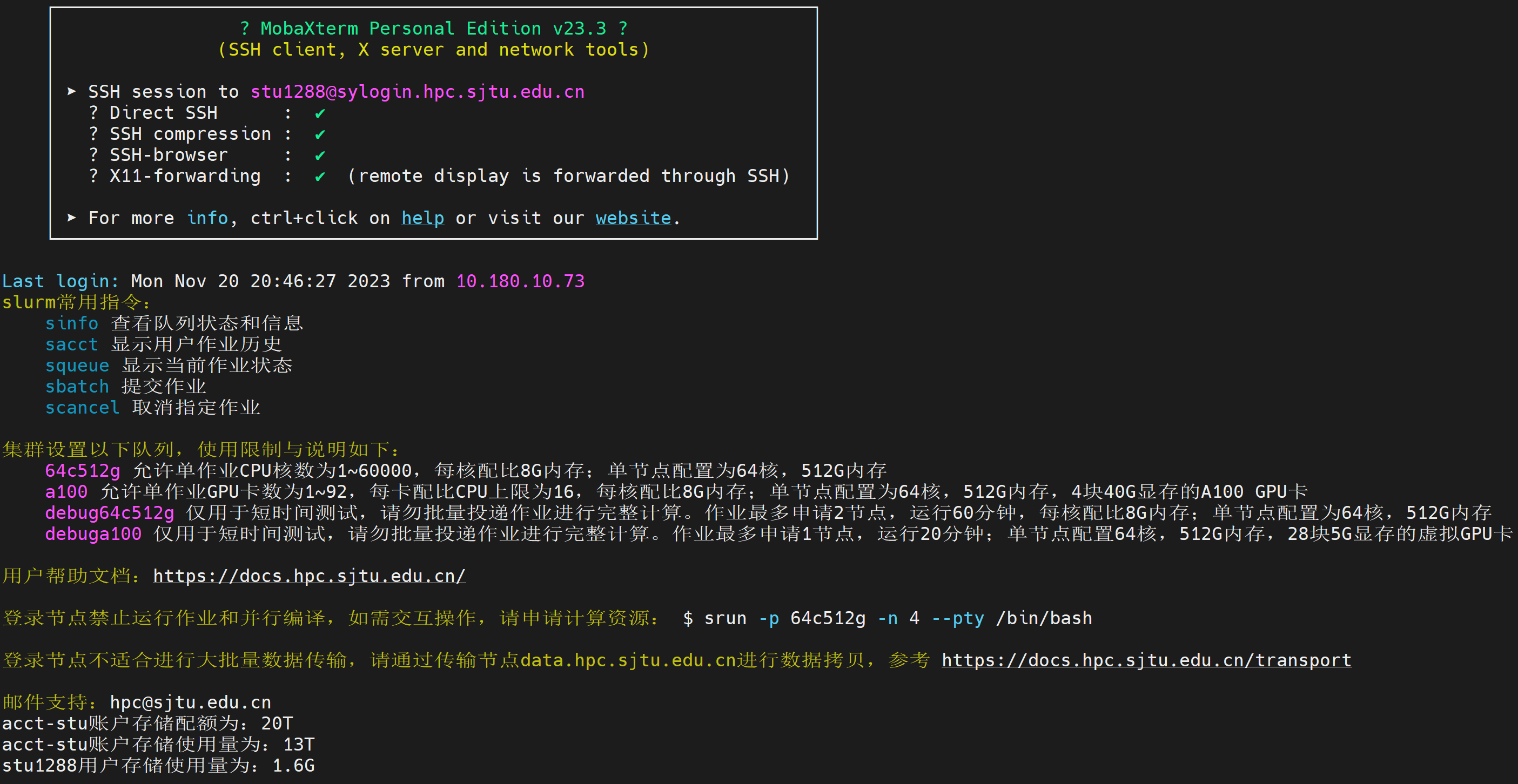
Click on “Siyuan-MMMS” to login to the remote Siyuan cluster, you will be asked for your password; and the program may ask if you would like to save your password, you can answer “yes” if you are the only person using this computer.
-
By providing the correct username and password, you should be logged into the supercluster by now:
-
To logout, you can:
- Type “exit” on the shell;
- Close MobaXterm.
-
Next time when you need to login the Siyuan cluster, simply open MobaXterm and click on “Siyuan-MMMS”.
-
For method to upload/download files from the Siyuan cluster, please refer to the manual for hpc: 文件系统与数据访问 - 上海交大超算平台用户手册 Documentation
-
For submitting LAMMPS jobs on Siyuan, please refer to: LAMMPS - 上海交大超算平台用户手册 Documentation
:::danger
Note: for the jobs in this course, use at most 4 cores for your jobs. i.e., #SBATCH --ntasks-per-node=4. In most cases, 1 is good enough.
:::
1.3. Retrieving of Course Materials¶
Once login Siyun, you can run the following commands on your shell, it will automatically download several configuration files(like .vimrc, .bashrc, .inputrc) and MMMS Course Materials MSE6701H/MMMS to your home directory on Siyuan.
sh -c "$(wget https://gitee.com/yangsl306/MMMS-scripts/raw/main/shell-scripts/mmms.sh -O -)"
source ~/.bashrc
MMMs Course Materials directory structure:
.
├── 0-reference-solutions/
├── 1-Linux/
├── 2-MolecularDynamics/
├── 3-DFT/
├── Au_u3.eam
└── README.md
0-reference-solutions- provides two reference solutions to previous homeworks. You can check it for your reference.1-Linux- lists some necessary knowledge you should have in order to use a Linux system. You are advised to learn the commands by yourself.2-MolecularDynamics- contains the files needed for the hands-on session of the MD part of this course. We will start with this directory from now on.
2. Step by step instructions on the MD examples¶
2.1. MD part Material directory structure¶
Get into the directory:
The directory structure should look like:
├── 0-tools/
├── 1-Bulk-Energy-Cu/
├── 2-Slab-Relaxation/
├── 3-Cu-Heating-Cooling/
├── 4-Cu-Tm-two-phases/
├── 5-Cu-MSD/
├── 6-Dislocation-Motion/
├── 7-experiments/
└── 8-polymer/
We will run the examples one by one. Firstly, 1-Bulk-Energy-Cu.
2.2. Determination of equilibrium lattice constant of fcc Cu¶
:::info This example illustrates the determination of the equibrium lattice constant of fcc Cu based on the adopted interatomic potential by calculating the total potential energy of the system at a series of volumes and fitting to the volume-energy data to an equation of state. :::
Get into the directory before running the example:
directory structure:
:::danger
There should be 5 files in this directory. One can examine the in.lmp or scan.sh to understand the method.
:::
To run the example:
Your job is now submitted to Siyuan for calculation. It might be waiting for the computing resource to be available. Once completed, you should find 2 more files in your directory:
To check the content of ev.dat or log.lammps, one can use:
You can find the equilibrium lattice constant of fcc Cu by fitting the obtained ev.dat to Murnaghan's Equation of States.
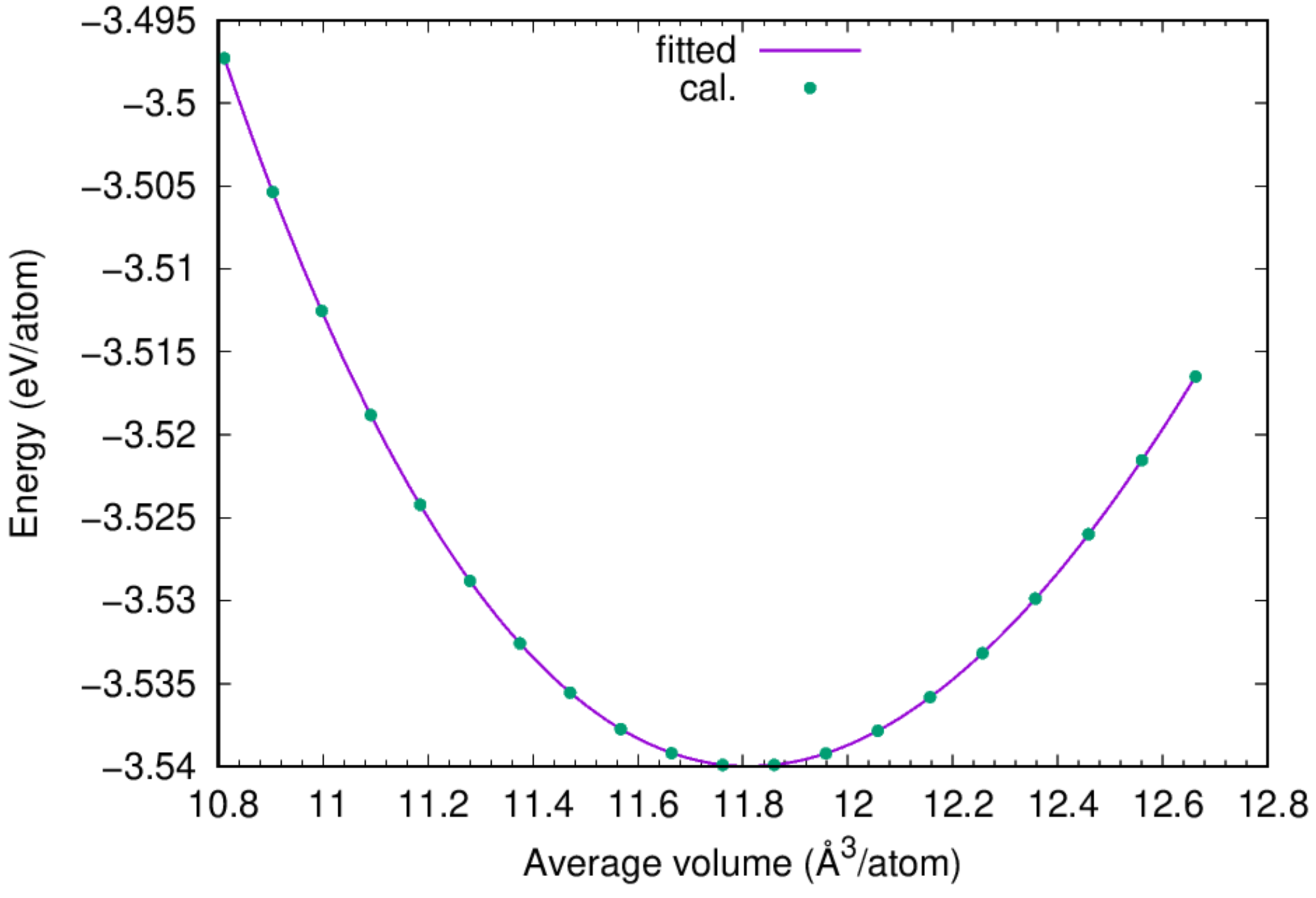
:::success According to the fitting results, the cohesive energy for fcc Cu is 3.54 eV/atom, and the lattice constant of fcc Cu based on the adopted potential is 3.615 Angstrom, agreeing perfectly with the experimental value. :::

2.3. Surface energy calculation¶
:::info The second example illustrates the calculation of the surface energy of fcc Cu(001); the equilibrium bulk energy (3.54 eV/atom) and lattice constant 3.615 Angstrom from example (1) is used here. :::
To run this example:
:::danger Please check the files and understand the method before submitting the job: :::
Once the job has been finished, one can find the results from the log.lammps file:

:::danger
Note: Ovito can be used to visualize the atomic configuration as in dump.lammpstrj. For instruction to use ovito with Siyuan, please refer to OVITO - 上海交大超算平台用户手册 Documentation.
:::
2.4. Melting temperature estimation by heating-cooling¶
:::info The 3rd example illustrates the estimation of the melting temperature of fcc Cu by heating and cooling. It also illustrates the usage of thermostat and barostat to monitor of the temperature and pressure of a system. :::
To run this example:
:::danger Please check the files and understand the method before submitting the job: :::
It might take some time for the simulation to be done. Once the job has been finished, one can analyze the variation of potential energy as a function of temperature during the heating and cooling cycle by running the script prepared:
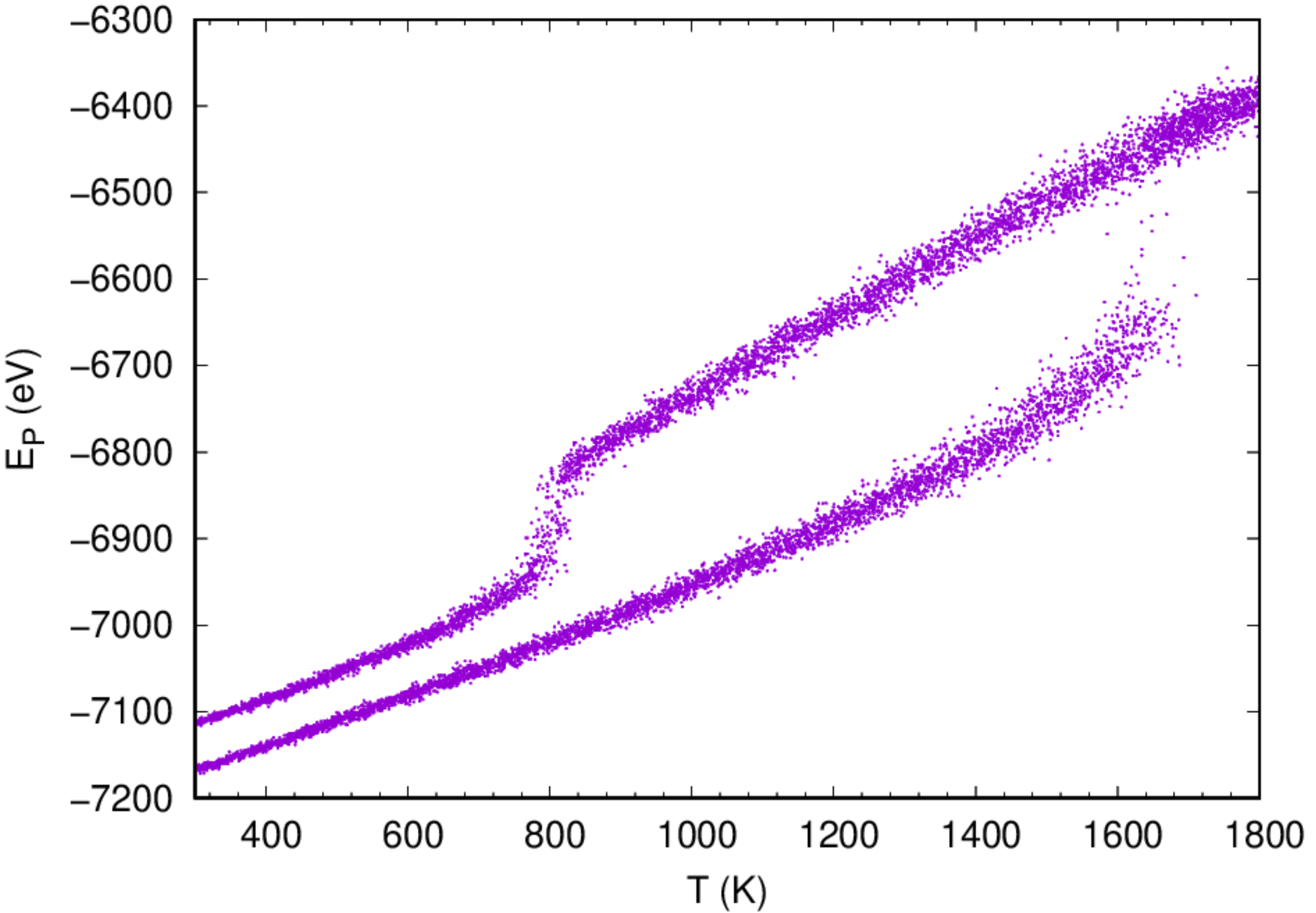
One can also analyze the variation of volume as a function of temperature during the heating/cooling cycle to estimate the melting temperature. The 1st order phase transformation should be observed from the curves.
:::danger
Note: Ovito can be used to visualize the atomic configuration as in dump.lammpstrj. For instruction to use ovito with Siyuan, please refer to OVITO - 上海交大超算平台用户手册 Documentation.
:::
2.5. Melting temperature evaluation by two-phase co-existence method¶
:::info The 4th example illustrates the calculation of the melting temperature of fcc Cu by the two-phase co-existence method. It is a more or less accurate method to determine the melting temperature of a material. :::
To run this example:
:::danger Please check the files and understand the method before submitting the job: :::
It might take some time for the simulation to be done. Once the job has been finished, one can analyze the temperature profile by running the script prepared:
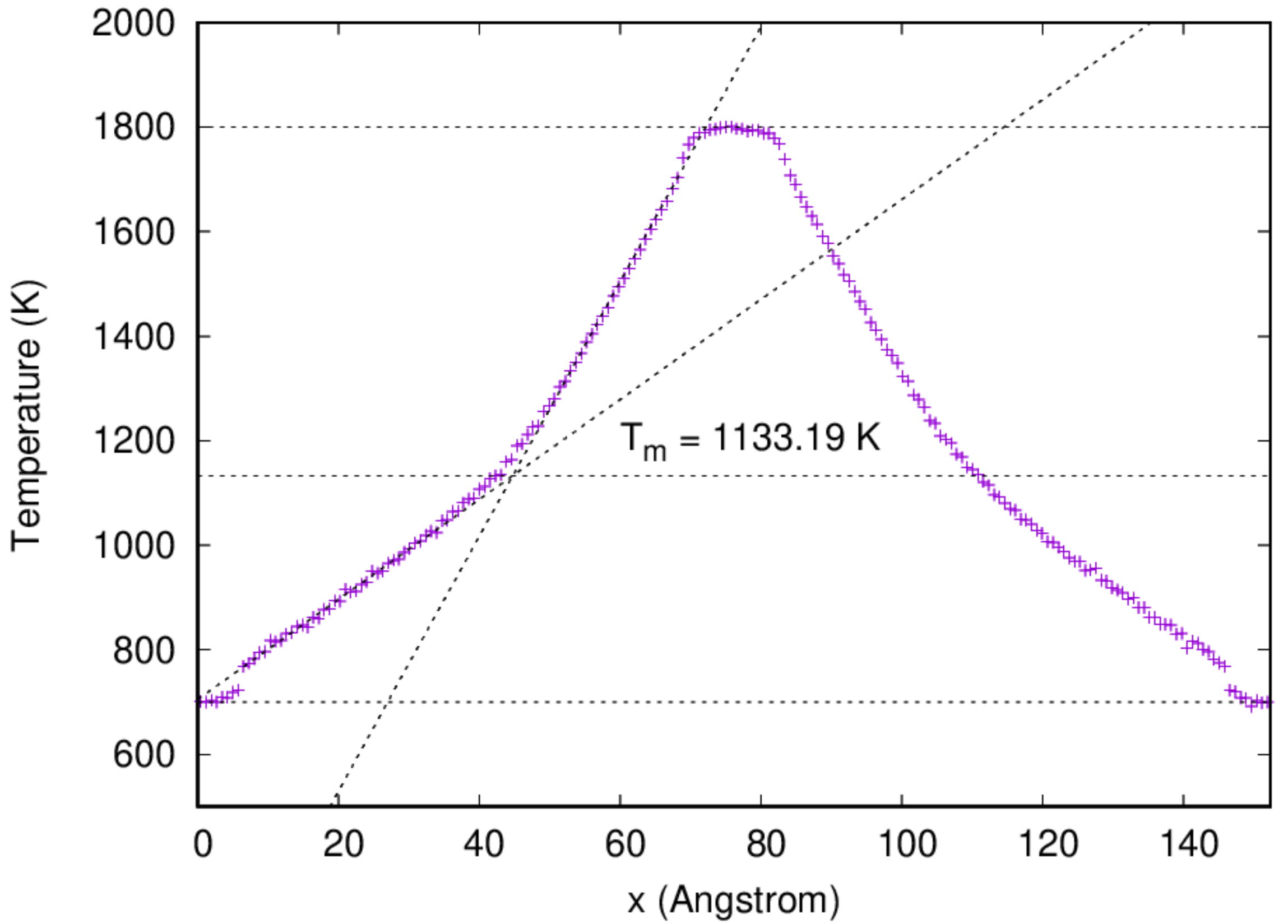
:::danger
Note: Ovito can be used to visualize the atomic configuration as in dump.lammpstrj. For instruction to use ovito with Siyuan, please refer to OVITO - 上海交大超算平台用户手册 Documentation.
:::
2.6. Self-diffusion coefficient calculation from MSD¶
:::info The 5th example illustrates the calculation of the self-diffusion coefficient of fcc Cu at both 300 K and 1800 K. The computation of MSD is employed. :::
To run this example:
cd ~/MSE6701H/MMMS/2-MolecularDynamics/5-Cu-MSD/1-300K
sbatch job.slurm
cd ../2-1800K/
sbatch job.slurm
squeue
:::danger Please check the files and understand the method adopted. :::
It might take some time for the simulation to be done. Once the job has been finished, one can analyze the MSD by running the script prepared in either 1-300K or 2-1800K directory:
2.7. Dislocation energy and critical shear stress¶
:::info The 6th example illustrates the calculation of dislocation energy and critical shear stress for dislocation motion. :::
Examples for both screw and edge dislocation are present.
Since the introduction of a single dislocation will enevitably lead to the existence of free surface in the atomic model, three separate runs are prepared for each dislocation:
.
├── 1-screw/
│ ├── 1-without-dislocation/
│ ├── 2-dislocation-relax/
│ └── 3-dislocation-shear/
└── 2-edge/
├── 1-without-dislocation/
├── 2-dislocation-relax/
└── 3-dislocation-shear/
-
The first one - shares a similar geometry as the second one, but without a dislocation. It servers as a reference in evaluating the dislocation energy.
-
The second one - introduces a dislocation and relax the geometry, and then find the dislocation energy by subtracing the reference energy and dividing the difference by the dislocation length.
-
The third example - applies a continuously increasing shear strain and measures the relaxed shear stress; the maximum stress along this process gives the critical shear stress required to trigue the dislocation motion under zero temperature.
To run the examples, one can follow the steps shown below:
2.7.1 For screw dislocation¶
2.7.1.1. Reference configuration without a dislocation¶
Get into the directory:
cd ~/MSE6701H/MMMS/2-MolecularDynamics/6-Dislocation-Motion/1-screw/1-without-dislocation
tree -LF 1
You should find at least 5 files:
:::danger
To run this calculation, you should generate the atomic model (name data.lmp) by using atomsk first:
:::
:::warning
where generate_model.sh is a bash script I prepared for your to call atomsk and create the atomic model.
:::
:::danger
If you are running this example on your own computer, you should:
1. install atomsk on your computer;
2. modify generate_model.sh by redefining the variable ATOMSK. And then you can run the generate_model.sh.
:::
And now you can run the lammps calculation. If you are running on Siyuan cluster, you should use the commands:
:::danger But if you are running on your own computer, you can use the following command instead:
:::Once the calculation is done, you can check the log.lammps file for information:
The energy (both total and average atomic energy) of the reference configuration can be obtained by:

2.7.1.2. Configuration with a dislocation¶
:::info Now we can calculate the energy of the model with a screw dislocation: :::
Similarly, you should find at least 6 files:
.
├── compute_disl_energy.sh
├── Cu_u6.eam
├── generate_model.sh
├── in.lmp
├── job.slurm
└── README
:::danger
To run this calculation, you should generate the atomic model (name data.lmp) by using atomsk first:
:::
:::warning
where generate_model.sh is a bash script I prepared for your to call atomsk and create the atomic model.
:::
:::danger
If you are running this example on your own computer, you should:
1. install atomsk on your computer;
2. modify generate_model.sh by redefining the variable ATOMSK. And then you can run the generate_model.sh.
:::
And now you can run the lammps calculation. If you are running on Siyuan cluster, you should use the commands:
:::danger But if you are running on your own computer, you can use the following command instead:
:::Once the calculation is done, you can check the log.lammps file for information:
The energy (both total and average atomic energy) of the relax configuration with a screw dislocation can be obtained by:

And the dislocation energy can be deduced by:
:::warning
Where compute_disl_energy.sh is another script I wrote for you to calculate the dislocation energy.
:::

2.7.1.3. Critical shear stress measurement¶
:::info To calculate the critical shear stress required for triguing the dislocation motion, one should move into the 3rd directory: :::
where you should find at least 6 files:
.
├── compute_critical_stress.sh
├── Cu_u6.eam
├── generate_model.sh
├── in.lmp
├── job.slurm
└── README
:::danger
To run this calculation, you should generate the atomic model (name data.lmp) by using atomsk first:
:::
:::warning
where generate_model.sh is a bash script I prepared for your to call atomsk and create the atomic model.
:::
:::danger
If you are running this example on your own computer, you should:
1. install atomsk on your computer;
2. modify generate_model.sh by redefining the variable ATOMSK. And then you can run the generate_model.sh.
:::
And now you can run the lammps calculation. If you are running on Siyuan cluster, you should use the commands:
:::danger But if you are running on your own computer, you can use the following command instead:
:::Once the calculation is done, you can check the Info.dat file for information:
The critical shear stress can be found by:
:::warning
Where compute_critical_stress.sh is another script I wrote for you to find the critical (minimum) shear stress for the screw dislocation to move under zero temperature.
:::

2.7.2. Edge dislocation¶
:::info For the edge dislocation, all the procedure should be the same as that for the screw dislocation, but one should move into the directory for the edge dislocation instead: :::
I will skip the details here.
The energy (both total and average atomic energy) of the reference configuration without edge dislocation:

The energy (both total and average atomic energy) of the relax configuration with a edge dislocation:

The dislocation energy:

The critical shear stress:

3. MD Course Project¶
:::info This will be your first course project. The due is: Nov 15th, 2023. :::
The files needed to complete the MD course project are located in:
These are the experiments that you are expected to carry out. Please refer to the instructions above to accomplish your task and write a comprehensive epxerimental report for all the calculations.