ASE 使用¶
约 1350 个字 722 行代码 5 张图片 预计阅读时间 14 分钟
介绍¶
-
atomic simulation environment (ASE):一系列用于设置、操作、运行、可视化及分析原子模拟的工具和 Python 模块
-
ASE 可支持的构型文件格式:File input and output — ASE documentation
-
ASE 通过
Calculators为不同的计算代码(DFT/MD)提供接口,Calculators与核心Atomsobject 和 ASE 中的许多可用算法一起使用;支持的Calculators:

-
ASE 版本 Release notes:查看版本更新细节
-
注意事项:
- ASE 网站中的源代码参数及注释与安装的 Python package 源码会有不一致的地方,写脚本还是以 pacakge 的源码为准
- 无直接计算弹性常数的模块
- 很多变量的类型是
np.ndarray
参考资料¶
-
ase 教程(内容较详细):ASE tutorials;源码:GitHub - ASE-Workshop-2023/tutorial
-
ase tutorial:ASE_tutorial.ipynb
-
ase md 模拟:GitHub - PythonFZ/ase_md_example
-
弹性性质计算的 ASE 接口(弹性常数、EOS、声速;感觉一般):GitHub - jochym/Elastic: A module for ASE for elastic constants calculation.
-
基于 PAW 和 ASE 的 DFT code:GPAW: DFT and beyond within the projector-augmented wave method — GPAW
-
GitHub - AlexBoucherr/ASExVASP: A serie of script to perform calculations on VASP using the ASE
-
GitHub - jkitchin/dft-book: A book on modeling materials using VASP, ase and vasp
-
ase 结构 2D 和 3D 渲染:GitHub - chrisjsewell/ase-notebook: Highly configurable 2D (SVG) & 3D (threejs) visualisations for ASE/Pymatgen structures, within the Jupyter Notebook.
-
GitHub - superstar54/x3dase: X3D for Atomic Simulation Environment
-
ase symmetry 教程(内容一般):GitHub - ajjackson/ase-tutorial-symmetry: Tutorial notebook for symmetry features in ASE
-
lab5 有 ase NEB 计算:labutil/samples at master · bkoz37/labutil · GitHub
-
原子建模之ASE篇_哔哩哔哩_bilibili(主要生成纳米结构)
-
CompMatBook/Chapter04/4_2_辅助建模软件ASE生成纳米结构.ipynb at main · stanfordbshan/CompMatBook · GitHub
-
内容一般(能带计算 ASE Python 代码部分可参考): GitHub - WMD-group/ASE-Tutorials: Examples of using the Atomic Simulation Environment
安装¶
使用¶
CLI¶
# 开启 ase 补全(只适用于 bash,zsh 不行)
ase completion >> ~/.bashrc
# 列出 ase 可识别的构型文件格式
ase info --formats
# 列出 ase 的 calculators 以及是否被安装
# 3.22.1 与 3.23.0 版本的输出格式有区别
ase info --calculators
# 构型转换
ase convert -i vasp -o xyz -f -v POSCAR structure.xyz
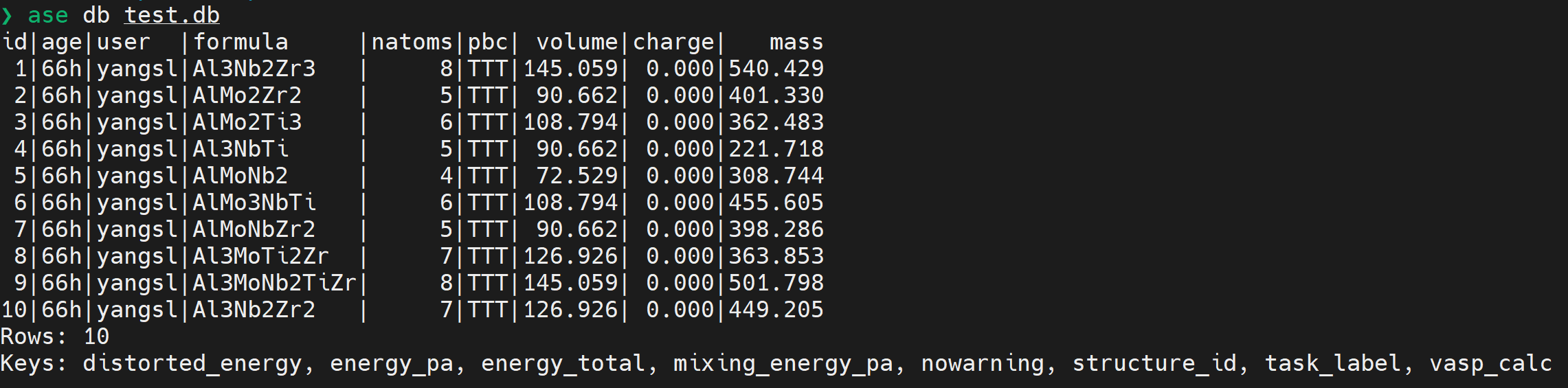
# 查看 db 文件内容 推荐
ase db test.db
-L N # 只显示前 N 行
--offset N # 跳过前 N 行
--show-keys # 显示所有 keys
--show-values key1,key2,... # 显示 key 的值;value 为数值时,只显示首尾值,如 energy_pa: [-9.184..-5.855]
其他¶
-
ase 缺陷计算 - 寻找最优的超胞形状:Tools for defect calculations — ASE documentation
-
interface 构建(较简单情况):Interface building - Manipulating atoms — ASE documentation
# 振动分析
from ase.vibrations import Vibrations
from ase.md.verlet import VelocityVerlet
# 简易元素周期表绘制
# reference: https://wiki.fysik.dtu.dk/ase/gallery/gallery.html
from ase.utils.ptable import ptable
atoms = ptable()
atoms.write("ptable.png")
常用模块¶
ase.atoms¶
-
Atom 和 Atoms 是 ASE 的两个基本 Object, Atoms 由 Atom 构成
-
Atoms 本质上是 Atom 的 list,可以使用标序的方式来查看 Atom
-
需添加 calculator 才能使用的 methods:Adding a calculator
from ase.atoms import Atoms
# 属性
symbols # 化学式;list() 得到原子对应化学符号列表
positions # 原子位置;笛卡尔坐标
cell # 基矢
cell[:] # 基矢;np.ndarray
cell.array # 同上
cell.cellpar() # 晶格参数(常数 + 角度)
cell.lengths() # 晶格常数
cell.angles() # 晶格角度
numbers # 原子对应原子序数
pbc # 周期性边界条件
constraints # 获取约束信息(原子 x y z 轴固定信息)
info # 给 Atoms 设置信息;dict;可用于写入 extxyz 格式文件
# (一般)方法
todict() # 将原子信息写入 dict
copy() # 拷贝
wrap() # 已施加 PBC 时,可将胞外原子移至胞内;下面的 wrap 参数同
rattle() # 随机移动原子(位置)
write() # 写入构型格式文件
edit() # 交互式修改(ASE GUI)
pop() # 删除原子
translate() # 平移原子位置
make_supercell() # 构建超胞(可通过此将六方转正交胞)
rotate() # 旋转(可绕轴旋转)
center() # 在指定轴两端各添加真空层并移至该轴中心
# 参数
vacuum # 真空层厚度
axis # 指定轴
# 方法;主要分为获取和设置;部分获取方法与属性的功能相同
get_xxx()
set_xxx()
# get 方法
get_pbc() # 周期性边界条件
get_cell() # 基矢
get_volume() # 体积
get_masses() # 原子对应原子质量;np.ndarray
get_atomic_numbers() # 原子对应原子序数;np.ndarray
get_positions() # 笛卡尔坐标;wrap 参数默认为 False
get_scaled_positions() # 分数坐标;wrap 参数默认为 True
get_chemical_formula() # 化学式
get_chemical_symbols() # 化学符号列表
get_distance() # 两原子间的距离
get_distances() # 第 i 个原子与给定原子列表间的距离
# get 方法;需添加 calculator 才能使用的方法
get_potential_energy() # 总能量;单位 eV
get_forces() # 每个原子的受力;单位 eV/Å
get_stress() # 应力张量;单位 eV/Å^3;voigt 参数
get_potential_energies() # 每个原子的能量
get_stresses() # 每个原子的应力张量
get_properties() # 获取性质,如 ["energy", "forces", "stress"]
get_total_energy() # 总能量
get_magnetic_moment() # 磁矩
get_magnetic_moments()
# set 方法
set_chemical_symbols() # 设置元素符号;可用于置换元素
set_constraints() # 施加约束;通常需结合 ase.constraints 的 FixAtoms 函数使用,直接设置 False/True 或 0/1 的列表无效果
- 晶体结构常用变量获取
from ase.atoms import Atoms
from ase.formula import Formula
atoms: Atoms
# 原子数
natoms = len(atoms)
# 元素种类数
nelements = len(set(atoms.get_chemical_symbols()))
# 化学式
formula = atoms.get_chemical_formula()
# 成分 {'Al': 5, 'Ti': 1}
composition = Formula(formula).count()
# 构型中某一元素的浓度
concentration = atoms.get_chemical_symbols().count("Pd") / natoms
# 原子对之间的最小、最大距离
import numpy as np
# 可考虑最小影像准则
distances = atoms.get_all_distances()
print(np.unique(distances)[1]) # 最小距离
print(np.unique(distances)[-1]) # 最大距离
# 判断 cell 是否为正交胞
angles = atoms.cell.angles()
is_orthogonal = all(abs(a - 90) < 1e-5 for a in angles)
- 其他用法
from ase.atoms import Atom, Atoms
atoms = ...
# 删除某一元素
del atoms[[atom.index for atom in atoms if atom.symbol == "H"]]
# 删除原子(形成空位)
del atoms[0]
# 添加原子(形成间隙缺陷)
atoms.append(Atom("H", position=...))
# 交换原子位置
positions[[0, 1]] = atoms.positions[[1, 0]]
# 生成纳米线
def make_wire(spacing: float = 2.5, box_size: float = 10.0) -> Atoms:
wire = Atoms(
"Au",
positions=[[0.0, box_size / 2, box_size / 2]],
cell=[spacing, box_size, box_size],
pbc=[True, False, False],
)
return wire
atoms = make_wire()
ase.build¶
- 结构建模
Bulk 晶体结构¶
from ase.build import bulk
# 原胞
atoms = bulk("Al", "fcc", a=4.05)
# 单胞 cubic=True
atoms = bulk("Al", "fcc", a=4.05, cubic=True)
# 支持的 crystalstructure 参数值
sc # 简单立方
bcc # Nb
fcc # Al
hcp # Mg
diamond # Si
bct # Body-Centered Tetragonal
rhombohedral # 菱形
orthorhombic # 正交
rocksalt # NaCl
cesiumchloride # CsCl
fluorite, caf2 # CaF2
zincblende # 闪锌矿 ZnS
wurtzite # 纤锌矿 ZnS
# crystal 构建
# 方式 1;简单晶体构建
from ase.build import bulk
# 方式 2;手动构建
from ase.atoms import Atoms
# 方式 3;利用空间群构建
from ase.spacegroup import crystal
# 超胞
# 方式 1
supercell = atoms * 2
# 方式 2
supercell = atoms * (2, 2, 2)
Surface 表面模型¶
-
无法枚举出具有不同表面终端的所有表面(建议还是通过 Material Studio 或 pymatgen surface 模块或 atomsk 构建含该表面坐标的单胞,进而查看原子层数)
-
常见晶体结构(BCC、FCC、HCP、Diamond)的常见表面(100、110、111)构建函数,支持吸附位点('ontop', 'bridge', 'hollow' 等)
-
指定面指数切表面(比 pymatgen surface 模块好用)
-
注意事项:
- fcc100、fcc110、bcc100、hcp10m10、diamond100 总是返回正交胞
- fcc111、bcc110、bcc111、hcp0001 可返回非正交胞和正交胞两种
- fcc211 只返回正交胞;diamond111 只返回非正交胞
- root surface 是什么含义?
from ase.build import ...
# 给定构型,沿指定面指数切表面
atoms = ...
s = surface(
lattice=atoms,
indices=(0, 0, 1),
layers=1,
vacuum=5.0,
)
# 参数
lattice # Atoms object
indices # 密勒/面指数
layers # 指最小一个完整单元的 slab,而非具体的原子层数
vacuum # z 方向两端添加真空层
# FCC 结构常见的 (100)、(110)、(111) 面
fcc100
fcc110
fcc111
fcc211 # 设置有要求
# BCC 结构常见的 (100)、(110)、(111) 面
bcc100
bcc110
bcc111
# Diamond 结构常见的 (100)、(111) 面
diamond100
diamond111
# HCP 结构常见的 (0001) 面
hcp0001
hcp10m10 # size 设置有要求;m 表示负号
mx2 # MoS2 二维材料的六方结构
graphene # 单层石墨烯
# 纳米带结构
graphene_nanoribbon # 石墨烯纳米带
# tube 结构
nanotube # 纳米管
# 示例
atoms = fcc100(
symbol="Cu",
a=3.615,
size=(10, 10, 16),
vacuum=30.0,
orthogonal=True,
)
# 通用参数
symbol # 元素符号
a # 晶格常数
vacuum # z 轴两端添加真空层
size # 指的是层数?
orthogonal # 是否转换成正交胞
- 其他
from ase.build import add_vacuum, sort
add_vacuum() # 添加真空层;单独使用该函数时,返回值为 None,即无效果
sort() # 按照元素符号排序生成新的 Atoms object
# reference: https://andyhox.github.io/2024/08/07/Learn-VASP-from-pymatgen-13/
# molecule 模块支持通过输入常见的分子化学式构建对应的分子结构
# 查看 ase 支持的分子种类
from ase.build.molecule import extra
from ase.collections import g2
print(g2.names)
print(extra.keys())
ase.visualize¶
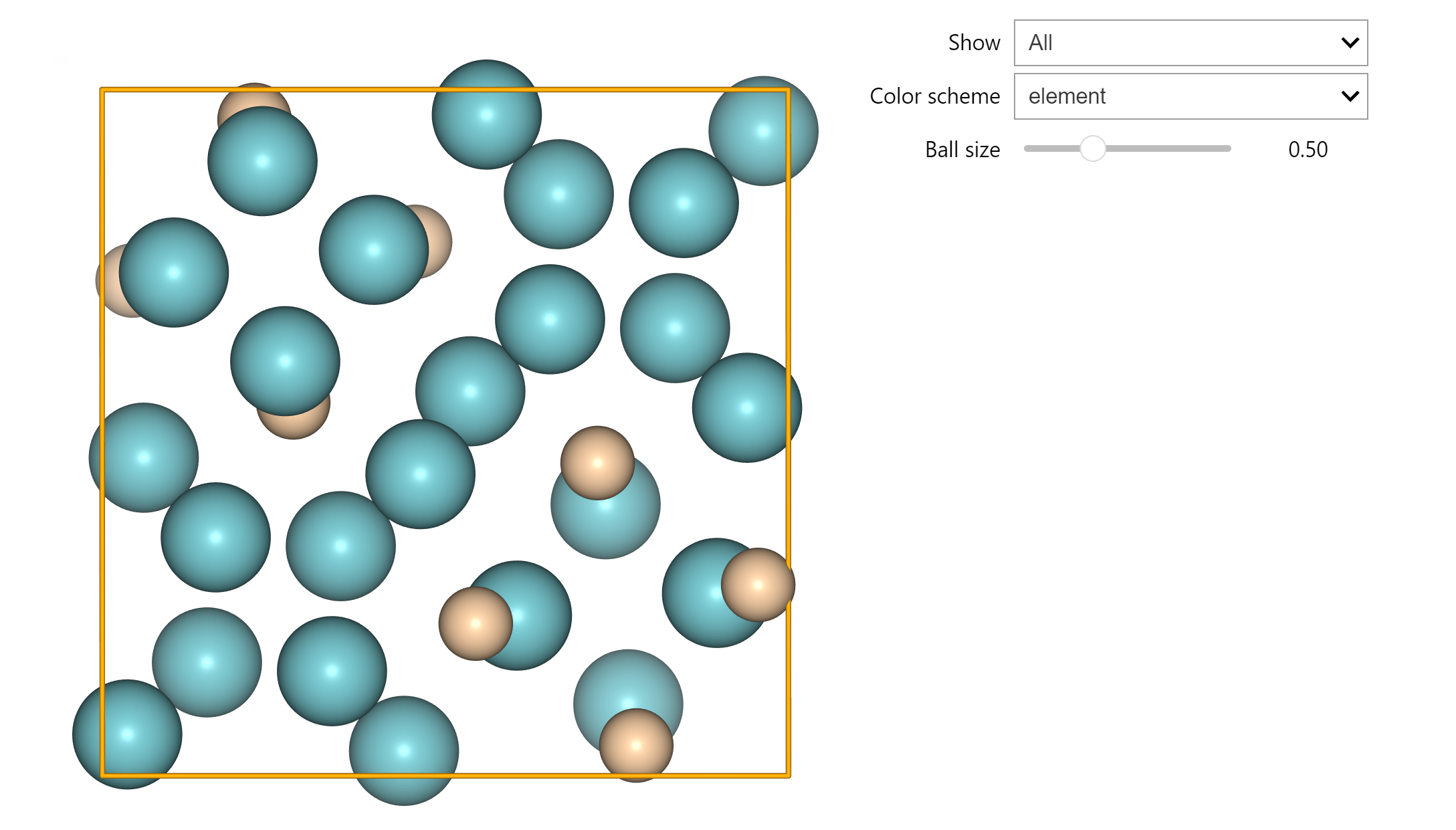
- 构型可视化(建议在 Jupyter Notebook 中使用)
# 方式 1
from ase.visualize.plot import plot_atoms
plot_atoms(atoms)
# 方式 2
from ase.visualize import view
view(atoms, viewer="ngl")
- nglview 组件效果图:
ase.cell¶
- 基矢/点阵
from ase.cell import Cell
# 生成基矢
cell = Cell.fromcellpar([3.31, 3.31, 3.31, 90, 90, 90])
cell[:] # 3x3 矩阵
# 方法
lengths() # 点阵长度
angles() # 点阵夹角
get_bravais_lattice() # 获取布拉维点阵
bandpath() # 能带路径
reciprocal() # 倒易点阵
niggli_reduce() #
minkowski_reduce() #
# 属性
volume # 体积
orthorhombic # 是否正交
rank # 秩
ase.spacegroup¶
- 空间群
from ase.spacegroup import Spacegroup, crystal, get_spacegroup
get_spacegroup() # 获取 Atoms 的空间群信息
crystal() # 类;通过空间群构建晶体结构
spg = Spacegroup(152) # 类;查看给定空间群的对称性信息
# 属性
symbol # 空间群符号
no # 空间群序号
scaled_primitive_cell # 基矢
# 方法
equivalent_sites() # 查看等同原子坐标
ase.lattice¶
- 点阵
有生成 graphene 和 graphite 模块:ase/lattice/hexagonal.py · master · ase / ase · GitLab
Bravais lattices — ASE documentation
ase.io¶
-
构型文件读入、写出;可读入压缩格式文件内容;可支持的格式很多;File input and output — ASE documentation
-
部分构型文件格式只读 (R) 或只写 (W) 或可读入写入多帧构型 (RW+) 数据
# 常用构型文件格式 读写情况
vasp # RW
vasp-out # R+
vasp-xdatcar # RW+
vasp-xml # R+
xyz # RW+
extxyz # RW+
xsd # RW
xtd # RW+
lammps-data # RW
lammps-dump-text # R+
cube # RW
cif # RW+
espresso-in # RW
espresso-out # R+
gpumd # RW
-
模块中的
read()函数可自动识别文件格式 -
将 OUTCAR 每个离子步信息转换成 extxyz 格式:Convert VASP OUTCAR to extxyz file for NequIP input · GitHub
-
写法一:在
read()、write()函数中指定format参数,即具体构型文件格式
from ase.io import read, write
from ase.data import atomic_numbers
# read() 函数
read(
filename=...,
index=...,
format=...,
**kwargs
)
# 参数
filename # 输入文件名
index # 读取单帧/多帧数据;int, slice, str 类型
format # 指定文件格式
**kwargs # 其他参数需参考具体的格式文件函数参数写法
# index 写法
index=0 # 第一帧构型
index=-2 # 倒数第二帧
index=':' # 所有帧
index=slice(None) # 同上
index='-3:' # 倒数三帧到最后
index=slice(-3, None) # 同上
index='1::2' # 偶数帧数据
index=slice(1, None, 2) # 同上
# write() 函数
write(
filename=...,
images=...,
format=...,
append=...,
**kwargs
)
# 参数
filename= # 输出文件名;"-" 表示标准输出
images= # 单个 Atoms 或 Atoms 列表
format= # 指定文件格式
append= # 是否写入多帧数据
**kwargs # 其他参数需参考具体的格式文件函数参数写法
# 保存为 VASP POSCAR 格式
write(
output_vasp_fn,
images=structure,
format="vasp",
direct=True,
sort=True,
vasp5=True,
ignore_constraints=Fasle,
)
# 参数
direct # 笛卡尔坐标/分数坐标
sort # 按照元素的字母顺序对原子进行排序
vasp5 # 以 VASP5+ 格式写入
ignore_constraints # 是否忽略约束(固定原子坐标信息)
# 保存为 LAMMPS data 格式
lammps_data_fn = ...
ele_list = ["Si", "Nb"]
write(
lammps_data_fn,
images=atoms,
format="lammps-data",
specorder=ele_list,
units="metal",
atom_style="atomic",
)
# 参数
specorder # 指定 atom type 顺序;默认按照元素符号字母排序
# 读取 LAMMPS data 格式
atoms = read(
lammps_data_fn,
format="lammps-data",
units="metal",
style="atomic",
Z_of_type={
1: atomic_numbers["Si"],
2: atomic_numbers["Nb"],
},
)
# 参数
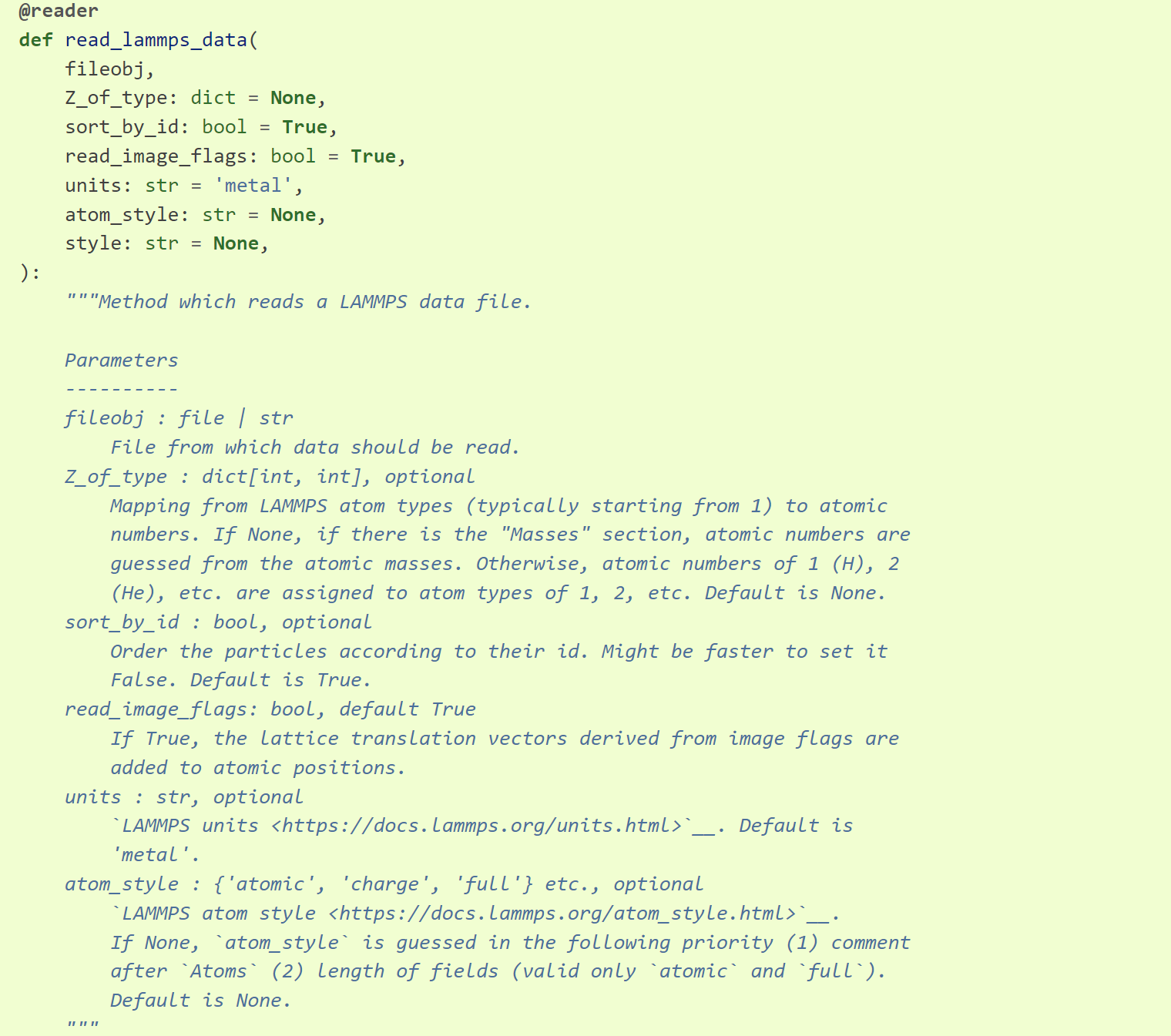
Z_of_type # dict,键为 type 编号,值为对应的元素原子序数;若为 None,有 Masses 信息,会根据其猜测原子序号,否则 type 编号对应的元素原子序数默认为 H He ... 等
# 将 OUTCAR 中每个离子步信息写入 extxyz
atoms_list = read("OUTCAR", index=":")
write(
extxyz_fn,
atoms_list,
format="extxyz",
append=True,
)
- 写法二:从
ase.io中导入具体构型文件格式的模块及其函数
# LAMMPS data 格式
from ase.io.lammpsdata import read_lammps_data, write_lammps_data
# LAMMPS dump 格式
from ase.io.lammpsrun import read_lammps_dump_text
# VASP POSCAR 格式
from ase.io.vasp import read_vasp, write_vasp
# VASP 输出文件格式 OUTCAR、XDATCAR、vasprun.xml
from ase.io.vasp import read_vasp_out, read_vasp_xdatcar, write_vasp_xdatcar, read_vasp_xml
# Material Studio xsd 格式
from ase.io.xsd import read_xsd, write_xsd
- 删除/重置 extxyz 文件中已有的 forces 和 info 信息:Issue with write_xyz (#276) · 议题 · ase / ase · GitLab
atoms = ...
del atoms.arrays["forces"] # 删除 forces 信息
atoms.info = {} # 添加 energy 等信息
# 从 extxyz 文件只能通过 get_potential_energy() 直接获取能量
# 而无法通过 get_forces() get_stress() 获取力、应力信息
xyz_fn = "train.xyz"
atoms_list = read(xyz_fn, index=":", format="extxyz")
atoms = atoms_list[0]
energy = atoms.get_potential_energy() # 可以
forces = atoms.get_forces() # 不可以;会报错
stress = atoms.get_stress() # 不可以;会报错
forces = atoms.arrays["forces"] # 可以
stress = atoms.info["stress"] # 可以
virial = atoms.info["virial"] # 可以
ase.constraints¶
- 施加约束
from ase.constraints import FixAtoms, FixedLine, FixedPlane
atoms = ...
# 按照 原子类型或坐标 对原子坐标轴进行固定
c = FixAtoms(mask=atoms.symbols == "Cu")
c = FixAtoms(mask=atoms.positions[:, 2] < 1.0)
# 允许沿 y 方向移动(固定 x、y 坐标轴)
c = FixedLine(
indices=[atom.index for atom in atoms],
direction=[0, 0, 1],
)
# 允许 yz 面移动(固定 x 坐标轴)
c = FixedPlane(
indices=[atom.index for atom in atoms],
direction=[1, 0, 0],
)
atoms.set_constraint(c)
ase.eos¶
- 执行 EOS 计算并拟合,得到体模量
from ase.eos import calculate_eos
from ase.units import kJ
from ase.atoms import Atoms
calc = ...
atoms: Atoms = ...
atoms.calc = calc
eos = calculate_eos(atoms, trajectory="XXX.traj")
v, e, B = eos.fit()
print(B / kJ * 1.0e24, "GPa")
- 根据 E-V 数据进行 EOS 拟合,获取平衡能量,体积和体模量;并绘制 EOS 拟合曲线
from ase.eos import EquationOfState
from ase.units import kJ
# eos 参数: birchmurnaghan murnaghan birch vinet
eos = EquationOfState(volumes, energies, eos="birchmurnaghan")
v0, e0, B = eos.fit()
print(f"v0 = {v0:.3f}")
print(f"e0 = {e0:.3f}")
print(f"B = {B / kJ * 1.0e24:.1f} GPa")
ax = eos.plot()
ax.set_title(label=None)
ase.db¶
-
db 文件
-
db.select(sort)中的sort为 含 key 的 str,含-时,降序
from ase.db import connect
from ase.db.row import AtomsRow
db_fn = ...
db = connect(db_fn) # 连接 db 文件
db.metadata = {...} # 给 db 添加元数据
len(db) # 获取 db 文件中存储的结构数目
db.count() # 同上
db.count("vasp_calc=Yes") # 添加 selection 筛选条件
# 若不存在,写入 empty row 并返回整数 id
id = db.reserve(key1=value1, key2=value2, ...)
# 筛选 id<=5 的所有结构
# selection 可以是 id 或其他 AtomsRow 中的 key
# 注:字符与符号之间不能有空格
for row in db.select("id<=5"):
...
# 筛选 id>=5, id<=10 的所有结构
for row in db.select("id>=5, id<=10"):
...
# 单个 AtomsRow
row = db.get(id=10) # id 从 1 开始
row._keys # 获取 AtomsRow 的 keys
row.key_value_pairs # 获取 AtomsRow 的 key_value_pairs
row.vasp_calc # 根据 key 获取 value
atoms = db.get_atoms(id=10) # 单个 Atoms
# 将 db 中的 AtomsRow 的结构和数据写入到其他 db 文件
db_output_fn = "..."
db_output = connect(db_output_fn)
for row in db.select("id<=10"):
key_value_pairs = row.key_value_pairs
data = row.data
# 将 AtomsRow 转化成 Atoms
atoms = row.toatoms()
db_output.write(
atoms=atoms,
key_value_pairs=key_value_pairs,
data=data,
)
ase.optimize¶
- 优化器(优化算法)
from ase.optimize import QuasiNewton # 等于 BFGSLineSearch
from ase.optimize.bfgs import BFGS
from ase.optimize.fire import FIRE
from ase.optimize.lbfgs import LBFGS, LBFGSLineSearch
from ase.optimize.mdmin import MDMin
ase.data¶
- 元素周期表中的元素相关数据
from ase.data import ...
# 大多都是 list、dict 或 np.array 的类型
# 第一个元素是 X,空位或让后续元素信息获取从 1 正常开始的含义
chemical_symbols # 元素符号
atomic_numbers # 原子序号
atomic_names # 元素英文全称
covalent_radii # 共价半径
cohesive_energies # 内聚能
reference_states # 基态
atomic_masses # 相对原子能量
vdw_radii # 范德华半径
ground_state_magnetic_moments # 基态磁矩
ase.units¶
ASE 中的物理单位,电子伏特 eV、埃 Å,开尔文 K 和原子质量单位定义为 1.0
from ase.units import Bohr, Hartree, eV, kJ, mol, GPa
# 能量: 1 eV = ... kJ/mol = ... Hartree = ... Ry
print(1 / (kJ / mol))
print(1 / Hartree)
print(1 / Ry)
# 长度单位
print(Bohr)
# 应力单位
print(1 / GPa)
ase.phasediagram¶
-
相图绘制(2 维,3 维):Phase diagrams and Pourbaix diagrams — ASE documentation
-
没有 pymatgen 对应的模块功能丰富且绘图效果好看
ase.cluster¶
- 纳米颗粒/团簇
ase.geometry¶
- rdf 计算
from ase.geometry import ...
# geometry 模块中的函数
get_duplicate_atoms() # 获取重复原子;可删除
is_orthorhombic() # 检查 cell 是否正交
permute_axes() # 扰动坐标轴
wrap_positions() #
# geometry 模块中的 Analysis 类
from ase.geometry.analysis import Analysis
ana = Analysis()
# 属性
images
nImages
nl # 近邻列表
all_bonds
all_angles
all_dihedrals
unique_bonds
unique_angles
unique_dihedrals
# 方法
get_bonds()
get_angles()
get_dihedrals()
get_rdf() # 计算 RDF(可计算 partial rdf)
ase.neb¶
from ase.neb import NEB
from ase.optimize import BFGS
images = ...
neb = NEB(images, k=0.1)
# IDPP 插值
neb.idpp_interpolate(fmax=0.1, optimizer=BFGS, steps=1000)
ase.md¶
-
ASE 中的 MD 相关算法
-
ASE 中的两种常见控压(压浴)方法:Parrinello-Rahman、Berendsen
-
使用 ASE md 模块执行 MD 示例:
from ase.md import MDLogger
from ase.md.npt import NPT
from ase.md.langevin import Langevin
from ase.md.nptberendsen import NPTBerendsen
from ase.md.nvtberendsen import NVTBerendsen
from ase.md.velocitydistribution import MaxwellBoltzmannDistribution, Stationary
ase.phonons¶
- 有限位移法计算声子谱
# 示例
from ase.phonons import Phonons
N = 25
ph = Phonons(atoms, calc, supercell=(N, N, N), delta=0.05)
ph.run()
ph.read(acoustic=True)
kpath = atoms.cell.bandpath(
[["G", "X", "U"], ["K", "G", "L", "W", "X"]],
npoints=100,
)
bs = ph.get_band_structure(kpath)
ase.filters¶
ase.neighborlist¶
- 近邻列表
ase.calculators¶
-
ASE 支持的 calculators:Supported Calculators — ASE documentation
-
ASE 原生 Python 实现的 caculators:EMT (effective medium theory),EAM,Lennard-Jones,Morse 和 HarmonicCalculator
VASP¶
-
设置 VASP 执行命令和赝势路径(在
~/.{bash,zsh}rc或在 Python 脚本中设置环境变量)
- ASE VASP Calculator 赝势不同泛涵目录命名
- 示例代码
from ase.calculators.vasp import Vasp
atoms = ...
calc = Vasp(
istart=0,
icharg=2,
encut=400,
ismear=1,
sigma=0.2,
lreal="Auto",
kpts=[5, 5, 5],
ivdw=12,
ediff=1e-05,
lwave=False,
lcharg=False,
net_charge=..., # 带电体系设置
)
atoms.calc = calc # 执行 VASP 计算
atoms.set_calculator(calc) # 同上,但写法过时
atoms.get_potential_energy() # 获取能量
from ase.calculators.vasp import Vasp
calc = Vasp(directory=".")
# 方法
read_convergence() # 是否收敛
read_results()
LAMMPS¶
- 示例代码
from ase.calculators.lammpsrun import LAMMPS
parameters = {
"pair_style": "meam/c",
"pair_coeff": ["* * library.meam Au Au.meam Au"],
}
files = ["library.meam", "Au.meam"]
calc = LAMMPS(parameters=parameters, files=files)
GPAW¶
- 示例代码
from gpaw import GPAW
from ase.calculators.emt import EMT
atoms = ...
calc = GPAW(mode='lcao', basis='dzp', txt='gpaw.txt')
atoms.calc = calc
opt = BFGS(atoms, trajectory='opt.traj')
opt.run(fmax=0.05)
设置单点能¶
# 设置单点能
from ase.atoms import Atoms
from ase.calculators.singlepoint import SinglePointCalculator
# 还可设置 forces、stress
results={"energy": -7.0}
calc = SinglePointCalculator(atoms, **results)
atoms.calc = calc
atoms.get_potential_energy()